

Hypertension (HTN) is defined as systolic blood pressure (BP) of 140 mmHg or higher or a diastolic BP of 90 mmHg or higher at the age of 20 years, and 160/95 mmHg at the age of 50 years[1]; however the Joint National Committee of World Health Organization on prevention, detection, evaluation and treatment of high BP have defined two stages of HTN in its 7th report. Stage 1 comprises systolic BP between 140-159 mmHg and diastolic BP between 90-99 mmHg; whereas stage-II with systolic BP >160 mmHg and diastolic>100 mmHg [2]. In the majority of cases, a specific underlying cause of HTN is not found and they are said to have essential HTN. Essential HTN is one of the major public health problems in many countries due to its high prevalence and its association with coronary heart disease (CHD), stroke, renal disease, peripheral vascular disease, and other disorders. [3]
Hypertension is a risk factor for the development of atherosclerosis, although the mechanisms have not been well elucidated. As the cellular and molecular mechanisms of the pathogenesis of atherosclerosis and the effects of hypertension are being more clearly defined, it becomes apparent that the two processes have certain common mechanisms. The endothelium is a likely central focus for the effect of both diseases. There is increasing evidence that atherosclerosis should be viewed fundamentally as an inflammatory disease.
Atherogenic stimuli such as hyperlipidemia appear to activate the inflammatory response by causing the expression of mononuclear leukocyte recruiting mechanisms. The gene for one of these, the vascular cell adhesion molecule-1, is controlled at least in part by transcriptional factors regulated by oxidative stress, which modifies the redox state of the endothelial cell. Alterations in the redox state of the arterial wall also may contribute to vascular smooth muscle cell growth. In a somewhat parallel fashion, there is evidence that hypertension may also exert oxidative stress on the arterial wall. This article reviews evidence that leads to the postulate that hypertension predisposes to and accelerates atherosclerosis at least in part because of synergy between elevated blood pressure and other atherogenic stimuli to induce oxidative stress on the arterial wall. [4]

Hypertension is not only a well-established cardiovascular risk factor but also increases the risk of atherosclerosis. Clinical trials have shown that, in the highest quintile of diastolic pressure, even with the added risks of high cholesterol and smoking, hypertension still contributes significantly to risk for atherosclerosis.[5] [6] In laboratory studies in which hypertension was induced in the Watanabe heritable hyperlipidemic rabbit, Chobanian, and his group [7] showed a synergistic effect that caused an intensification of atherosclerosis. In the total surface area of involvement of the abdominal aorta of these endogenously hypercholesterolemic animals, a dramatic enhancement of lesion formation accompanied the induced hypertension. The low-density lipoprotein (LDL) receptor in this rabbit strain has the same type of molecular defect as is found in familial hypercholesterolemia. Thus, both clinical and experimental data show that high (elevated) blood pressure enhances the development of atherosclerosis. In fact, atherosclerosis tends to occur only in those parts of the vascular system subjected to high pressure.
However, the mechanisms of this enhanced or synergistic effect are not yet well defined. We still do not understand the fundamental nature of atherosclerosis itself, although great progress has been made, especially recently, in understanding and developing some unifying hypotheses about the pathogenesis of the disease. This conceptual process, in turn, has made it easier to begin to develop mechanistic insights into the role of hypertension in exacerbating the atherosclerotic process.
Atherosclerosis and hypertension are distinct disease entities; everyone who has hypertension does not manifest extensive atherosclerosis, nor is atherosclerosis always or even usually accompanied by hypertension. The cardinal pathological features of atherosclerotic lesion development are (1) the presence of monocytes/macrophages and T cells, (2) their localization in large conduit or elastic arteries in areas of low shear stress, (3) proliferation and migration to the intima of medial smooth muscle cells, (4) the deposition of increased amounts of connective tissue, and (5) neovascularization.[8] Hypertensive arteries are thickened, and there may be increased smooth muscle cell mass and/or cell number and increased deposition of connective tissue.[9] In considering the interactions of the two diseases, it will probably be most useful to consider mechanisms or consequences that are common to both. The purpose of this review is to relate recently developed concepts of the pathogenesis of atherosclerosis to shared mechanisms in hypertension, with a focus on the molecular mechanism by which hypertension might facilitate the development or progression of atherosclerosis.
The Atherosclerotic Triad: Oxidation Stress, Inflammation, and Endothelial Dysfunction
Oxidative stress, endothelial dysfunction, and inflammation represent a triad serving as the foundation for the development and progression of the multi-step process of atherosclerosis. Among these exist important and complex interactions in which cause-effect relationships are often difficult to distinguish.

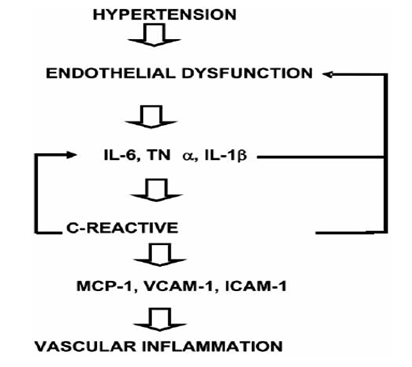
One of the earliest steps in the atherosclerotic process is endothelial dysfunction. Endothelial cells play a central role in the regulation of vascular function by controlling vascular tone, inflammation, and thrombosis through the production of numerous factors including nitric oxide (NO), inflammatory cytokines, pro-thrombotic agents, and anticoagulant factors. In a normal environment, there is a balance among secretions of these factors which maintains the integrity of the endothelial surface and ensures protection of the vessel. The presence of cardiovascular risk factors such as hypertension, diabetes, hypercholesterolemia, and smoking alters endothelial function. This endothelial dysfunction is characterized by a vasoconstrictor tone, increased leukocyte adhesion, and a prothrombotic stage. [10, 11, 12]
In these conditions, imbalance among endothelial factors initiates diverse events that play a pivotal role in the initiation and progression of the atherosclerotic process by initiating the formation of a fatty streak.
The main cause underlying endothelial dysfunction is a reduction in NO availability and subsequent reduction in all beneficial effects of this factor, which is not only a potent vasodilator but also inhibits leukocyte and platelet adhesion or modulates smooth muscle proliferation thus exerting antiatherogenic actions. These reduced NO levels involved, in addition to minor NO production, a higher NO breakdown by reactive oxygen species (ROS) which rapidly react with NO forming peroxynitrite, a highly reactive species that produces lipid peroxidation and protein nitration. [13]
In normal conditions, ROS (which includes various compounds such as superoxide anions, hydrogen peroxide, and hydroxyl radical) are produced in mammalian cells during energy production in the mitochondria by reducing oxygen during aerobic respiration. [14] In addition, a variety of enzymatic and non-enzymatic sources of ROS exist in vascular vessels as well as different tissues, among which can be included nicotinamide dinucleotide (phosphate) (NAD(P)H oxidase enzyme complex, xanthine oxidase, lipoxygenases, and cyclooxygenases. An uncoupling of NO synthase can also contribute to ROS production. [15]
Hypertension and other cardiovascular risk factors are frequently associated with oxidative stress since levels of its markers such as malondialdehyde, 8-isoprostane or 8-oxo- 7,8-dihydro-2′-deoxyguanosine is increased in both patients and models of hypertension. This seems to be a consequence of an imbalance between ROS production and antioxidant defense systems which become overwhelmed. This affirmation is based on the fact that an increase in NAD(P)H oxidase activity or expression has been reported with high blood pressure levels.
Mechanisms Involved in Tandem Hypertension – Inflammation
Although precise mechanisms that link inflammation and hypertension are not well established, changes in mechanical stress associated with hypertension as well as humoral factors involved in the development and complications of hypertension have been proposed Endothelium is exposed to mechanical stress created by blood flow and the cardiac cycle. The pulsatile nature of arterial blood flow in combination with a complex geometric vascular network configuration determines endothelial shear stress patterns that can be complicated by changes in hemodynamic forces associated with high blood pressure levels. [16] Endothelial cells are able to sense mechanical stress because they are equipped with numerous mechanoreceptors.
Their activation triggers a cascade of intracellular pathways that interact among themselves through cross-talk, especially the mitogen-activated protein kinase (MAPKs). All of these signaling pathways lead to phosphorylation of several transcription factors such as NF-kB which modulate the expression of mechanosensitive genes. These genes include ROS production from NADPH oxidase and other enzymes such as xanthine oxidase, as well as activation of modulators of cytoskeleton such as protein kinase C and Rho family small GTPases with subsequent structural changes in the arterial wall. NF-kB activation, at the same time, leads to an increased expression of several adhesion molecules (VCAM-1, ICAM-1, E-selectin), chemoattractant chemokines(MCP)-1, and pro-inflammatory cytokines such as TNF-α, and IL-1β Consequently, it not only favours an inflammatory process but also the development of atherosclerosis.

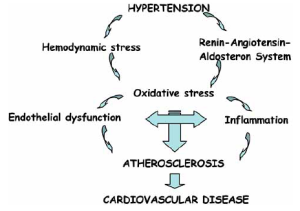
The renin-angiotensin-aldosterone system (RAAS) plays an important role in the regulation of the cardiovascular system through control of extracellular fluid volume, sodium balance, and functional and structural cardiac and vascular effects. In addition, RAAS overactivity is associated with the development of hypertension, atherosclerosis, left ventricular hypertrophy, and cardiovascular events such as myocardial infarction, stroke, and congestive heart failure. Angiotensin II, the main effector of the RAAS, is not only a vasoconstrictor factor but is also profibrotic and proinflammatory through its binding to the angiotensin type 1 (AT1) receptor. The proinflammatory effect of angiotensin II is mediated, in part, through the activation of NF-kB and subsequent production of a whole variety of inflammatory mediators. [17] This role has been clearly confirmed by the observation that angiotensin II antagonism results in a reduction of inflammatory markers.
Hypertension and hyperlipidemia exert many similar effects on the arterial wall. The increase in oxidative stress, a mechanism common to both conditions, may activate genes involved in generating an inflammatory response that, in the presence of hyperlipidemia, leads to the formation of atherosclerotic plaque. There is a great deal of interest in the use of antioxidants in the treatment of atherosclerosis. The possibility that members of this class of compounds might also ameliorate hypertensive vascular injury deserves further investigation.
Scholarly References:
1. Vasan RS, Beiser A, Seshadri S, et al. Residual life time risk for developing hypertension in middle aged men and women; The Framingham Heart Study. JAMA 2002; 287: 1003-10
2. Chobanian M. The 7th Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure. JAMA 2003; 289: 2562-74.
3. Gong M, Hubner M. Molecular genetics of human hypertension. Clin Sci 2006; 110: 315-26.
4. Alexander, W.R., Hypertension and the Pathogenesis of Atherosclerosis. Hypertension. 1995; 25: 155-161
5. Stamler J, Neaton JD, Wentworth DN. Blood pressure (systolic and diastolic) and risk of fatal coronary heart disease. Hypertension. 1989;13(suppl I):I-2-I-12.
6. Kannel WB, Neaton JD, Wentworth D, Thomas HE, Stamler J, Hulley SB, Kjelsberg MO. Overall and coronary heart disease mortality rates in relation to major risk factors in 325,348 men screened for the MRFIT: Multiple Risk Factor Intervention Trial. Am Heart J. 1986;112:825-836.
7. Chobanian AV, Lichtenstein AH, Nilakhe V, Haudenschild CC, Drago R, Nickerson C. Influence of hypertension on aortic atherosclerosis in the Watanabe rabbit. Hypertension. 1989;14:203-209.
8. Griendling KK, Alexander RW. Cellular biology of blood vessels. In: Schlant RC, Alexander RW, eds. Hurst’s The Heart. 8th ed. New York, NY: McGraw-Hill Publishing Co; 1994:31-45.
9. Chobanian AV. Vascular effects of systemic hypertension. Am J Cardiol. 1992;69:3E-7E. 6 Doyle AE. Hypertension and vascular disease. J Cardiovasc Pharmacol. 1992;19(suppl 5):S7-S10.
10. Baeuerle PA, Baltimore D. Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-kappa B transcription factor. Cell. 1988;53:211-217.
11. Alexander RW. The coronary ischemic syndromes: relationship to the biology of atherosclerosis. In: Schlant RC, Alexander RW, eds. Hurst’s The Heart. 8th ed. New York, NY: McGraw-Hill Publishing Co; 1994:1021-1031.
12. Berk BC, Alexander RW. Biology of the vascular wall in hypertension. In: Brenner BM, ed. Pathophysiology of Renal Disease. Philadelphia, Pa: WB Saunders. In press.
13. Alexander RW, Hennigar RA, Griendling KK. Pathogenesis of hypertension: vascular mechanisms. In: Braunwald E, ed. Atlas of Heart Diseases: Atherosclerosis—Risk Factors & Treatment. Philadelphia, Pa: Current Medicine; 1995:4.1-4.16.
14. Crawford DW, Blankenhorn DH. Arterial wall oxygenation, oxyradicals, and atherosclerosis. Atherosclerosis. 1991;89:97-108.
15. Nakazono K, Watanabe N, Matsuno K, Sasaki J, Sato T, Inoue M. Does superoxide underlie the pathogenesis of hypertension? Proc Natl Acad Sci U S A. 1991;88:10045-10048.
16. Cunningham KS, Gotlieb AI. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest 2005; 85: 9-23.
17. Kranzhofer R, Browatzki M, Schmidt J, Kubler W. Angiotensin II activates the proinflammatory transcripcion factor nuclear factorkappaB in human monocytes. Biochem Biophys Res Commun 1999; 257: 826-8.











[…] Hypertension and the Pathogenesis of Atherosclerosis | Pharma Mirror […]