By Dr Ash Ramzan, founder and principal consultant, Woodley BioReg
During a pharmaceutical product’s lifetime, it’s widely known and accepted that manufacturing processes will drift within their specified ranges — typically due to equipment wear-and-tear and operator variance. To maintain the ‘validated state’, diligent producers spend a lot of time and effort in ensuring validation and periodic re-validation of processes to ensure that any process drift is accounted for and controlled.
The registration file — such as the Marketing Authorisation (MA) or Product Licence (PL) — may be considered as the equivalent to the manufacturing process, and from which parameters can often wander, a phenomenon described as ‘Registration Drift’. Typically, this occurs through the movement of personnel, poor or inadequate change control, and mergers and acquisitions.
Registration drift can often be exacerbated where products are manufactured by third parties, as technical and quality agreements generally don’t robustly address change control and communication between organisations can be poor. If left unchecked, this situation can quickly escalate – resulting in serious conformance errors, presenting a risk to patient safety and product efficacy.
Despite clear legal and ethical obligations to ensure that MAs are updated whenever there is a reportable change, such as in the manufacturing process — i.e. materials, equipment, tests, specifications etc. — Registration Drift is a known phenomenon that until recently has not been fully understood (or addressed).
How do I know if registration drift is occurring?
To begin to assess and understand the impact of potential differences between the registration file and the manufacturing documents, a gap analysis must be performed to compare differences between them. While there are several ways to tackle this issue, the general approach for review of MAs against plant records is summarised in Figure 1 below — highlighting many of the key stages and activities for licence conformance review.
It’s worth noting that to ensure a complete and robust conformance assessment of the registration documents, the registration documents should be compared against the manufacturing documents; as not all plant details will be documented in the MA, an evaluation of the plant documents against the licence alone will result in only a partial review.

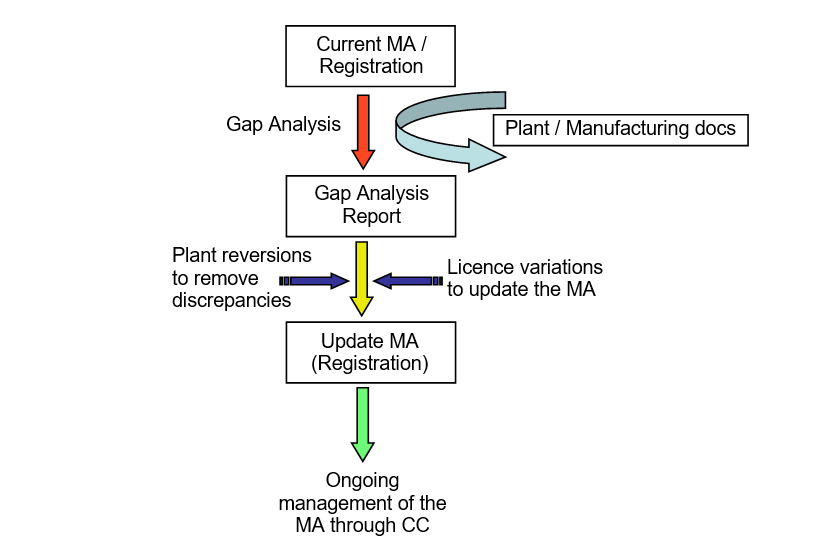
Following this comparison, a report documenting the differences should be prepared so that they can then be assessed and remediated in one of two ways. Firstly, in instances where it’s relatively straightforward to return to the registered details, the manufacturer may choose to revert its operations and effectively remove the discrepancy.
Alternatively, where reversion is not possible — such as if the equipment is no longer available or the methodology is no longer acceptable – the registration file must be updated through the submission of a licence variation. Both of these remediation actions must be performed under Change Control and will close the gap.
In generally, there are two main hurdles to overcome before rectifying conformance gaps; firstly, recognising that a problem exists, and secondly, knowing how to manage it without unnecessarily alerting the authorities and jeopardising a product’s future, or patient safety — the key to success for any conformance programme.
Acknowledgement that there is an issue is usually viewed as acceptance of the failure of one or more of the quality systems, and can potentially have wider implications on their management and implementation within an organisation. Interestingly, such ‘quality failures’ are also indicative of a well-developed quality system, with the ability to effectively self-audit to find discrepancies and manage them.
Resolutions may involve introducing a combination of possible short-term and long-term solutions to re-align the registered details with manufacturing operations. This can include giving due consideration to some of the sustainable solutions that a number of progressive pharmaceutical manufacturers have adopted to address this industry-wide problem.
The most important element of any long-term plan is the need to re-evaluate the purpose and scope of change control, with the re-implementation of this quality system to be inclusive of regulatory actions following a change in the manufacture, testing, or release of product. This effectively makes change control a single, end-to-end system applicable across the entire business, both operationally and geographically.
Do you have a problem?
From experience, there are a number of pointers known to have a direct impact on the degree of Registration Drift. These risk factors include:
- Whether there are a significant number of products that are more than around 15 years old.
- If products are manufactured by third parties that are not subject to robust technical/quality agreements.
- Whether a company has been subject to a merger or acquisition in the past.
- If there has been significant staff turnover, resulting in poor hand-over of responsibility and a lack of communication.
- Extensive use of contracted staff (as opposed to outsource service providers)
There are many other factors, but those listed above tend to by the typical culprits when Registration Drift has been identified. Given the potential complexity of products and supply chains, the question of whether this exists in a particular organisation is most effectively answered by undertaking a targeted evaluation of a small selection of MAs, with the answer becoming immediately clear.
What are the possible solutions?
There are several actions a company can take to address instances of Registration Drift. These are proactive and seeking to resolve potential issues and preferred over regulatory action where regulators will seek to implement enforced actions to assure patient safety; these are considered further below:
Proactive action
This is by far the best approach to addressing potential conformance issues. Often once a company has conducted a mini assessment, it quickly becomes apparent if there is Registration Drift present. From experience, there is always some degree of it regardless of the organisation, therapeutic area, or product type.
There are a number of fundamental steps to take to ensure that manufacturing details, tests, specifications, etc. are accurately captured within the MAs and remain aligned. These include:
- Resolving any apparent discrepancies
- Implementing measures to ensure that any errors remain fixed
- Performing periodic checks to ensure ongoing conformance
It’s important to emphasise that changes to registrations should not be avoided, but should be embraced as part of the product improvement and refinement process, while ensuring that products and registrations remain synchronised.
Following the completion of a gap analysis assessment, there needs to be one consistent procedure whereby MAs are maintained and managed. To enable this, a single repository of the registered information should be developed and kept up to date.
Regulatory action
Pharmaceutical manufacturers are subject to a variety of audits and assessments as part of a licensing authority’s obligations, including regular compliance audits. It’s not unusual for an auditor to ask for the product manufacturing details so that they can undertake an evaluation of the conformance between what is registered, and what is documented on site. Any observations will trigger a more detailed review of the complete registration file, and can include several penalties for the MA holder.
Registration Drift in product conformance is a real problem that exists to a lesser or greater extent in all pharmaceutical manufacturers. Many have already started a review programme, underlining the importance of this work, and some have completed them. There are a number of solutions, and selecting the path of least resistance should be based on specific circumstances and the products affected to minimise disruption.
Dr. Ramzan Bio:
With over 30 years’ experience in the biopharmaceutical and pharmaceutical industry, Dr. Ramzan is an expert in the development of drugs, biologics and medical devices / IVDs and provides consulting services to global corporations in Europe, USA, South Asia, and Australia.
He has provided regulatory bodies in the USA and the EU with dossiers to successfully register products including pharmaceuticals and biologics / vaccines and has successfully delivered a number of global regulatory and quality projects with household pharma names.
Dr Ramzan assists manufacturers in the management and implementation of regulatory conformance programmes for both pharmaceuticals and biopharmaceuticals, including biologics and vaccines.
He is also a fellow of TOPRA and editor of the Regulatory Rapporteur and a member of the British Pharmacopeia Working Party, which advises the government on the development of standards for biosimilar products.
Dr. Ramzan received his Ph.D in Protein Biochemistry and BSc. (Hons) in Biochemistry from the University of Liverpool.
About Woodley BioReg:
Woodley BioReg helps pharmaceutical, biopharmaceutical, healthcare, and medical device companies develop and manufacture quality products that are effective and safe for patients worldwide.
Providing best-in-class scientific advice and consulting services in regulatory affairs, quality, clinical/nonclinical, and project management for all stages of product development for 21 years, Woodley BioReg acts as the link between the global boards of health, regulatory agencies, and healthcare product manufacturers.